Structure Discrimination and Magnetic Properties of FeSnN with N =8–15
 Rivic, Filip_Structure Discrimination and Magnetic Properties of FeSnN with N =8-15
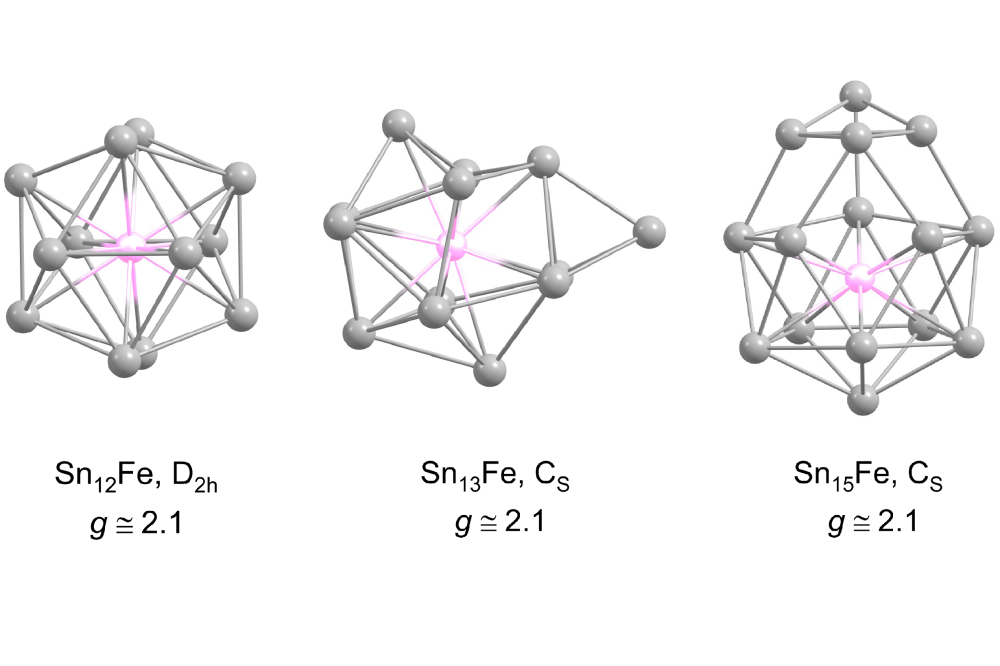
Rivic, Filip_Structure Discrimination and Magnetic Properties of FeSnN with N =8-15Figure 1: Predidcted ground state structures (GM) for SnNFe with N = 12, 13 and 15 at the PBE0/def2-TZVPP level of theory with their corresponding symmetry and calculated g-factors. Tin atoms are displayed as gray spheres while the iron atom is shown in pink.
Filip RivicEinleitung
In the framework of the Collaborative Research Center (CRC 1487), iron is studied as a substitute for rare-earth metals in environmentally friendly technologies, since they are toxic or obtained by disputable methods. For this, iron is required to be investigated in varying compositions in order to reveal its characteristics, often being tunable in nature. In this regard, we want to study the influence of tin atoms as ligands on the iron atom and investigate their intriguing magnetic properties. We could already synthesize iron-doped tin probes and generate molecular beams which could then be investigated in electric and magnetic beam deflection experiments. To further validate and interpret the electric and magnetic deflection profiles, we want to examine computationally the geometries and the magnetically-relevant properties comprising g-factors and spin-rotation coupling constants. The latter properties are also to be calculated for Pb12X clusters (X = Al, Ga, In) which are predicted to possess very high g-factors.
Methoden
Energetically-favored initial geometries were generated using a plane-wave Density Functional Theory-based (pw-DFT) Genetic Algorithm (GA) developed in our group. From the generated pool of structures candidates in the range of 1 eV relative to the energy of the energetically lowest-lying isomer are optimized on the PBE0/def2-TZVPP level of theory and verified by frequency analyses. The magnetic properties are then calculated with a relativistic Hamiltonian (ZORA).
Ergebnisse
The geometric structures of SnNFe with N = 8–15 were calculated within this project. For most of the cluster sizes only one isomer is found within the considered energy range of 0.1 eV which is assumed to be a conceivable energy range for our experimental setup. However, the found geometries cannot be assigned clearly to the experimental data. Further, we calculated the electronic g-factors of the clusters Sn12Fe, Sn13Fe and Sn15Fe to investigate the size-dependency of this property which allows good insight into the magnetic properties. Here, we could observe no such dependency, since these clusters all showed very similar g-factors of 2.1 slightly increased relative to the g-factor of a single electron (g ≈ 2).
Additionally, the magnetic properties of Pb12X clusters (X = Al, Ga, In) were calculated. Here, highly increased g-factors of about 4 were calculated for the Pb12Al and Pb12In clusters. In contrast to that, the g-factor of the Pb12Ga cluster is decreased to g = 1.6.
Diskussion
Since the iron atom has a spin multiplicity of S = 2, the calculation of the geometrical structures of SnNFe clusters is more complex than for the previous calculated S = ½ systems. This was also evident in the computing time needed to optimize one cluster structure on the higher level of theory. Single optimization steps needed more time than initially assumed. However, the used computing resources were still within the range of the estimation. The increased computing time is most likely due to the computational routine of the GA. Here, the spin multiplicity is not considered, since the cluster is treated as a unit cell of a solid to reduce the needed computational resources. However, the resulting initial geometric structures are not suited as good starting points for the further optimization and need much more time to converge. To circumvent this problem to either verify the calculated structures for the respective sizes or be able to calculate even larger clusters with more tin atoms, the initial structures have to be more accurate. This can be achieved by either introducing the spin multiplicity into the calculation routine of the GA or by choosing the initial structures without the GA by considering other already calculated open shell systems with higher spin multiplicity. The calculated magnetic properties indicate also that the cluster possess a considerable high zero-field splitting constant in the range of (10–30)cm-1.
Furthermore, the strongly increased g-factors in the Pb12Al and Pb12In cluster can be explained on the one hand by a very high spin-orbit coupling constant of Pb (three times higher than the one for tin) and on the other hand by the nonvanishing angular momentum in the contributing molecular orbitals. These molecular orbitals, however, do not contribute to the g-factor in the case of the Pb12Ga cluster. This can be understood by considering the d-block contraction in the gallium atom which leads to an icosahedral rather than a pyritohedral structre.