Modeling of Large Proteins in DNA Repair
 Modeling of large Proteins in DNA Repair
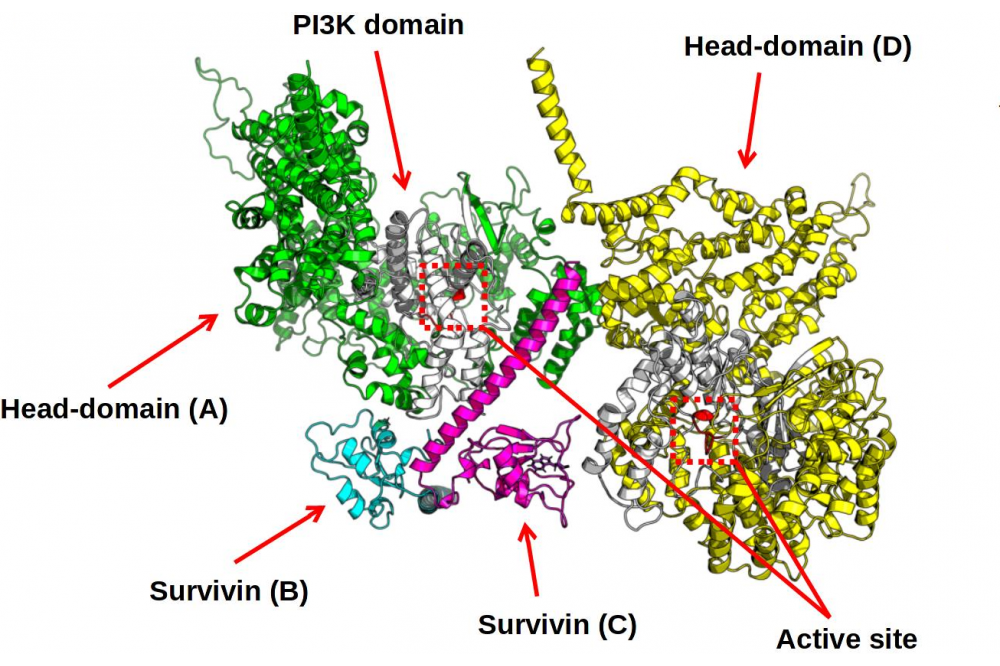
Modeling of large Proteins in DNA RepairFigure 1: Visualization of a DNA-PKcs - Survivin - DNA-PKcs heterotetramer structure.
Benjamin MayerEinleitung
DNA-PKcs is a gateway protein of the Non-Homologous End Joining DNA Repair Pathway. Which is the process of repairing DNA double strand breaks, by cutting away DNA until the break ends can be joint again. Besides being involved in a multitude of other processes, DNAPKcs binds to DNA break ends via a Protein complex called Ku70/80 hetero-dimer [1]. Together, these make up the DNA-PK complex, which may protect the break end until further proteins of the NHEJ machinery are recruited [1].
Understanding how to inhibit this protein by disabling its assembly or blocking its active site where crucial chemistry happens, as well as binding with other bio molecules, enables the in silico study of potential cancer treatments or how to enhance existing ones. Inhibiting NHEJ usually leads to an increased sensitivity of cells for radiation treatment and might make resistant tumors accessible to conventional chemo therapy again [2].
Here we aimed to study the interaction between DNA-PKcs and a certain cancer marker protein, i.e., Survivin, which lead to the discovery of a new Protein complex [3]. Complementary, the problem of finding a closed structure for the missing regions in the used DNA-PKcs Protein Structure has been investigated.
Methoden
A candidate for the interaction between the DNA-PKcs and Survivin had been produced using results from a previous Master Thesis [4] and a PhD Thesis [5], followed by protein-protein docking where only the so called head of DNA-PKcs, formed by the upper parts: the Kinase and Fat domain, had been used as docking partner for Survivin on a local cluster. Combining results from experiments carried out as part of a PhD Thesis [5] with these docking results a candidate complex of two DNA-PKcs heads connected by a single Survivin Protein, was selected. To verify that this indeed is a stable complex a molecular dynamics simulation for 200ns has been carried out on LB2.
To gain a closed model from the used structure 5LUQ, Rosetta Loop Remodeling has been used to gain one initially closed model. Here a relaxation of the region containing the ABCDE Phosphorylation pattern between residues 2500 and 2700 has been performed. Here a large ensemble of 100k structures has been produced. All of which have been filtered and then clustered by the Rosetta scores, for better visualization of the results.
Ergebnisse
A new heterotetramer structure made out of two DNA - PKcs bound together by Survivin has been found, as visualized in Figure 2. The Survivin Dimer seems to glue together the two DNAPKcs head regions by two different binding modes of the two identical Monomers making up the Survivin Dimer, with the PI3K region, colored in White respectively. The structure turned out to be stable in the molecular dynamics simulation, as verified by plotting the RMSD over time and seeing that the RMSD stabilizes after initial configurational changes.
The ten best found clusters from an ensemble of over 100k relaxation results has been visualized in Figure 3. Here the relaxed structure of the ABCDE Pattern is show in cartoon mode, while for the rest of the DNA-PKcs protein only the surface is colored indicating the different sub-regions.
Diskussion
The new-found heterotetramer can be used as basis for finding new small molecules inhibiting this specific interaction. For this, first results are published in the according PhD Thesis by Güllülü [5].
The generated ensemble of energy relaxed gap structures can be used for finding the actually whole structure. This is, e.g., possible by doing advanced sampling simulations starting from the lowest score structure found here. Of course, these results also need to be tested against real world structures and dynamics. In general, having a closed structure is necessary for a lot of computational modeling approaches, e.g., design of (peptide) binders, therefore motivating this task.