Investigation of Domain Switching Mechanics of Perovskite Ferroelectrics using Molecular Dynamics Simulations
 Investigation of Domain Switching Mechanics of Perovskite Ferroelectrics using Molecular Dynamics Simulations
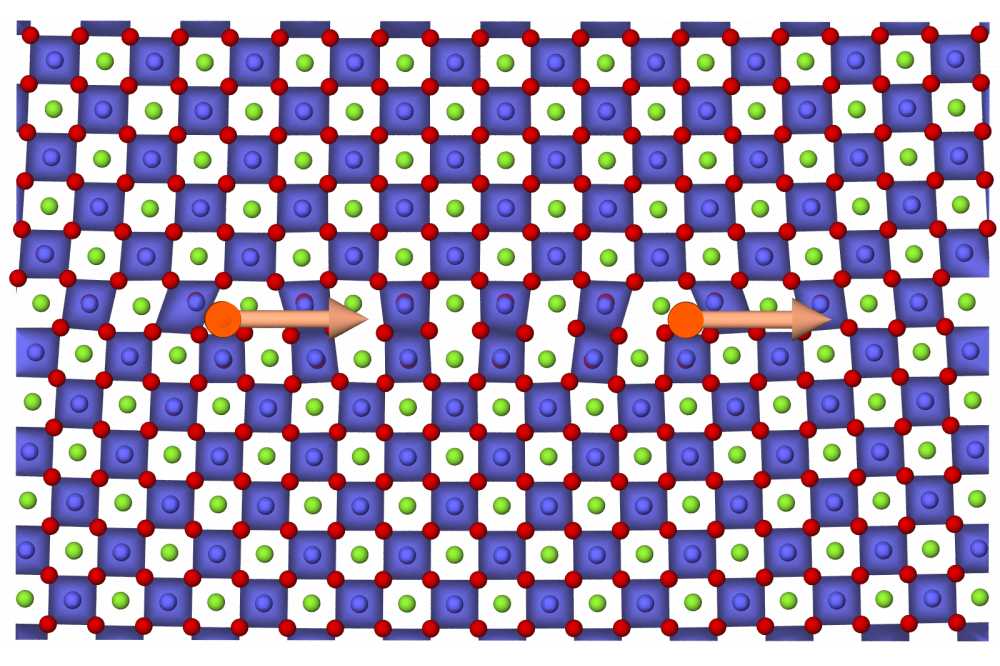
Investigation of Domain Switching Mechanics of Perovskite Ferroelectrics using Molecular Dynamics SimulationsFigure 1: Complex dislocation structure of a dislocation in a perovskite crystallattice. Oxygen (red) spans octahedra around the titanium atoms (blue) that are in the center of the strontium (green) cage. Dislocations and the corresponding Burgers vectors are indicated in orange. The stacking fault in between the two partial dislocations is apparent from the faulty connectivity of the octahedra.
Arne Klomp Investigation of Domain Switching Mechanics of Perovskite Ferroelectrics using Molecular Dynamics Simulations
Investigation of Domain Switching Mechanics of Perovskite Ferroelectrics using Molecular Dynamics SimulationsFigure 2: Elongated volume of barium titanate where the orientation of the local dipoles is color coded. A multi domain state is simulated in the tetragonal phase of barium titanate.
Arne KlompEinleitung
Ferroelectric ceramics with perovskite structure can be used in novel cooling applications based on the electrocaloric effect. The electrocaloric effect exploits the reversible switching of the ferroelectric microstructure of the material. In this study, we propose that defects in such a material, be it point defects or extended defects such as dislocations, influence the ferroelectric switching mechanism. Thus, we ultimately aim at understanding how defects can tune the electrocaloric response.
Methoden
The polarization and domain structure of ferroelectric compounds and especially the switching thereof are difficult to access by experimental means. This is due to the microscopic nature and the fast dynamics of the underlying physical phenomena. Therefore, we use simulations on the atomic (classical molecular dynamics or MD) and electronic (density functional theory, DFT) scale. They deliver accurate and detailed insights necessary to understand a material at the microscopic scale. In MD atoms are modeled as point masses that interact via effective potentials. Becoming increasingly complex these potentials allow description of many physical effects in ferroelectrics. For an efficient performance of the simulation we use the open-source computer code LAMMPS which works excellently on highly parallel computing clusters. In contrast, DFT also assumes atoms to be classical point masses but, additionally, includes a quantum mechanical treatment of the electron density. These simulations have the advantage of strongly increased accuracy at the cost of heavily increased computational load. Again an open-source solution, the computer code Abinit, implements an efficient framework for performing such calculations.
Ergebnisse
Prior to the current project, we have already gained significant expertise in the simulation of perovskite materials in the context of mechanical properties as well as modeling domain walls in MD simulations of a ferroelectric material. Regarding the mechanical properties of the perovskite ceramic strontium titanate, dislocation structures and their effects on plastic deformation have been studied in detail. Moreover, we have modeled domain walls in the ferroelectric compound barium titanate. The experience from these two fields, extended defects in perovskite materials and ferroelectric domains, are now combined. Consequently, we expect to show which type of defect can have distinct influences on the polarization microstructure and give guidelines to engineering the effect of defects. Engineering defects is, therefore, pursued as a route to tailor the material properties to achieve applicability in novel applications such as electrocaloric cooling.
Diskussion
The use of ferroelectric materials in modern highly specialized applications relies on the fundamental understanding of microscopic properties in the employed material. Atomistic and electronic structure simulations represent the optimal tool to gain an understanding of the underlying physical relationships. Making use of MD and DFT simulations we expect to relate the microscopic structure to macroscopic physical properties and, thus, aid in the design of ferroelectric materials.
Ausblick
Our research work is accompanied by research efforts throughout the Materials Science Department at TU Darmstadt that target the functional properties of ferroelectrics, anti-ferroelectrics and perovskites in general. We hope to pursue the perspective of a continuous improvement of these material classes on a large scale and make a contribution that enables bringing new functionalities to application.