Orbital-Dependent Exchange-Correlation Energy Functionals
Einleitung
In this phase of the project the exact exchange (EXX) of density functional theory was applied to both free-standing graphene and Si(111) slabs, using the plane-wave pseudopotential (PWPP) approach and a periodic repetition of the supercell containing the slab (in z-direction).
Ergebnisse
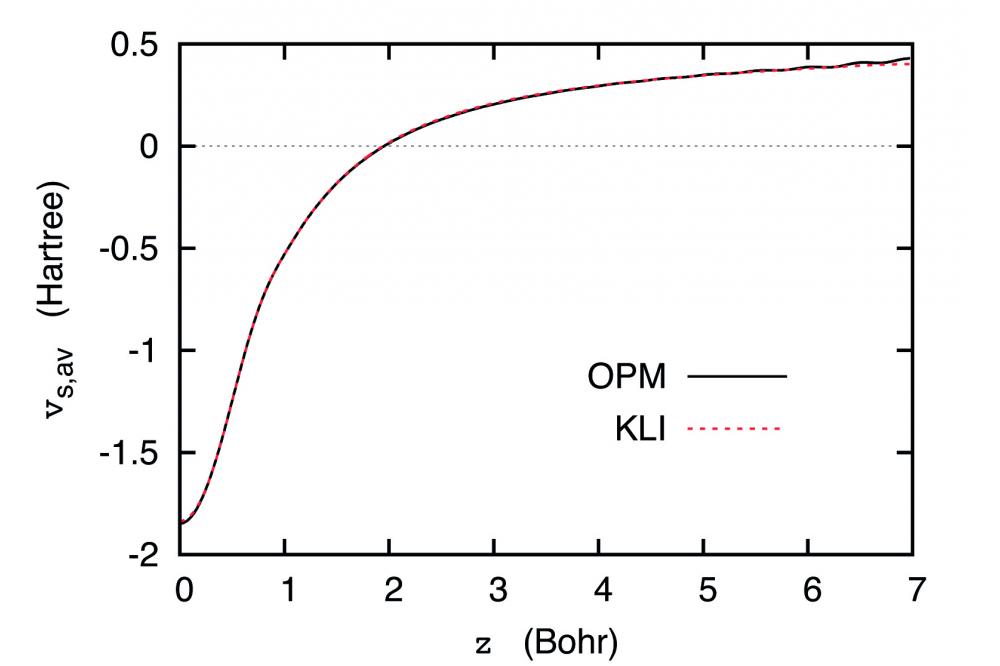
It was shown[1,2] that (i) EXX-PWPP calculations for slabs in supercell geometry are basically feasible (in spite of the high cut-off energy for the plane-wave basis required for an accurate description of the exponential decay of the Kohn-Sham states into the vacuum between neighboring slabs), (ii) the Krieger-Li-Iafrate approximation for the EXX potential[3] reproduces the exact EXX potential v_x obtained by solution of the EXX integral equation[4] (which is not feasible for large slab separation) quite accurately for moderate slab separation (Fig.1), (iii) the width of the vacuum required for a decoupling of the slabs is only slightly larger than in the case of the local-density approximation (LDA) (in spite of the longrange EXX functional), (iv) the corrugation of the EXX potential close to the surface of the slab is more pronounced than that of the LDA for v_x, (v) v_x shows an extended region, both far outside the surface of the slab and far from the middle of the vacuum region between the slabs, in which v_x behaves as -e^2/z, provided the width of the vacuum is chosen sufficiently large, and (vi) this intermediate -e^2/z behavior of v_x can be used for an absolute normalization of v_x and the total Kohn-Sham potential, which, in turn, allows the determination of the work function.
Diskussion
More recently, it was demonstrated in a Bachelor thesis[5] that the EXX predicts elastic constants for graphene which are somewhat closer to experiment than their LDA counterparts. Unrelated to the main theme of this project, PWPP calculations for those defects in hexagonal ice which trigger the melting process[6] were performed[7]. In particular, it was shown that density functional calculations yield a much higher barrier for the formation of the 5-7 defect than the phenomenological TIP4P potential[6].